-
Ion-Pair Dynamics upon Photoinduced Electron Transfer Monitored by Pump-Pump-Probe Spectroscopy
J.S. Beckwith, B. Lang, J. Grilj and E. Vauthey
Journal of Physical Chemistry Letters, 10 (13) (2019), p3688-3693


DOI:10.1021/acs.jpclett.9b01431 | unige:126819 | Abstract | Article HTML | Article PDF | Supporting Info
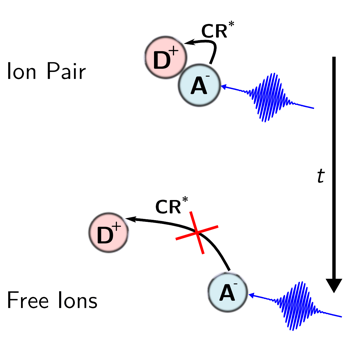
The excited-state dynamics of the radical anion of perylene (Pe) generated upon bimolecular photoinduced electron transfer (PET) with a donor was investigated using broadband pumpâââ‰â¬Åpumpâââ‰â¬Åprobe spectroscopy. It was found to depend on the age of the anion, that is, on the time interval between the first pump pulse that triggers PET and the second one that excites the ensuing Pe anion (Peâââ¬Ã¢Ã¢ââ‰â¬Å). These differences, observed in acetonitrile but not in tetrahydrofuran, report on the evolution of the PET product from an ion pair to free ions. Two photoinduced charge recombination pathways of the ion pair to the neutral Pe*(S1) + donor state were identified: one occurring in a few picoseconds fromÃâàPeâââ¬Ã¢Ã¢ââ‰â¬Å*(D1) and one taking place within 100âââ‰â¬Å200 fs fromÃâàPeâââ¬Ã¢Ã¢ââ‰â¬Å*(Dn>1). Both processes are sensitive to the interionic distance over different length scales and thus serve as molecular rulers.